

Un paciente de 54 años acudió a la consulta para valoración. Mientras realizaba el electrocardiograma se comprueba la posible existencia en el registro de una prolongación del intervalo QT (un parámetro del ECG que suele permanecer estable y constante). Ante el hallazgo se le pregunta si había en su familia antecedentes de muerte súbita relacionados con la natación, ruidos intensos, alarmas etc. Confiesa que su madre había muerto de forma repentina durante una tormenta. Confirmó que los testigos que le acompañaban en ese instante vieron como cayó desplomada tras oírse un gran trueno. El diagnóstico de síndrome QT largo familiar y sus posibles consecuencias estaba hecho.
En las sociedades avanzadas, la mayoría de las muertes súbitas se dan en personas de edad media o avanzada y son consecuencia de la enfermedad coronaria -cardiopatía isquémica en sus formas aguda o crónica. No obstante, un significativo porcentaje se da en sujetos aparentemente sanos y activos, y se relacionan con arritmias malignas producto de trastornos eléctricos cardíacos primarios de base congénita.
Los síndromes hereditarios de arritmias constituyen, por tanto, un conjunto de enfermedades de origen principalmente eléctrico que se dan en corazones estructuralmente normales y que se caracterizan por su base genética y su potencial letalidad mediante una susceptibilidad aumentada para desarrollar arritmias malignas. Actualmente se reconoce que hasta un tercio de los sujetos con muerte súbita y autopsia negativa se relacionan con estos trastornos. En algunos casos pueden detectarse cambios morfológicos en el ventrículo –que pasan inadvertidos frecuentemente si no se buscan de forma dirigida- y suelen carecer de trascendencia funcional y clínica. Finalmente, la miocardiopatía hipertrófica es una patología diferenciada tanto por su relativamente alta prevalencia como por aunar importantes cambios morfológicos y funcionales ventriculares junto a una marcada inestabilidad eléctrica.
Generalmente, este conjunto de patologías incide en personas no identificadas previamente como portadoras de tales anomalías eléctricas cardíacas. Como quiera que estos individuos tienen –salvo excepciones- un corazón estructuralmente sano y que la muerte se produce de forma inesperada en las primeras décadas de la vida y a veces relacionada incluso con el deporte, su impacto social es enorme. En esta revisión divulgativa, dividida en dos partes, se pretende repasar algunos fundamentos de los principales síndromes hereditarios arritmogénicos. En la primera parte se consideran las generalidades, los síndromes QT largo, y el síndrome de Brugada. En la segunda, las taquicardias ventriculares catecolaminérgicas, la miocardiopatía arritmogénica, la hipertrófica, y otros trastornos del ritmo de asociación familiar variable como ciertos tipos de fibrilación auricular o vías accesorias.
Origen de la inestabilidad eléctrica heredada del corazón
En la mayoría de estos trastornos hereditarios la arritmia se relaciona con la inestabilidad eléctrica de las células cardíacas, concretamente a nivel de sus canales iónicos, de ahí que frecuentemente se agrupan con el término canalopatías. Los canales iónicos cardíacos (Figura 1) son poros que modulan el paso de ciertos iones (moléculas con carga eléctrica) en determinadas circunstancias y que modifican el voltaje a nivel celular y en el conjunto del propio órgano. Son fundamentales en las células de corazón, cerebro, músculos y otros tejidos excitables. La funcionalidad de estas microestructuras debe ser perfecta para asegurar la completa estabilidad eléctrica del mismo, estando determinada por un conjunto de genes cuyas mutaciones causan estas enfermedades.
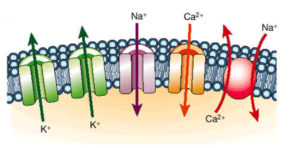
FIGURA 1. Los canales iónicos se construyen a partir de grandes proteínas que residen en las membranas de las células, siendo algunos de ellos selectivos para distintos tipos: sodio (Na), potasio (K), calcio (Ca), etc, y pudiendo ser activados por diferentes estímulos. Estas estructuras son responsables del flujo transmembrana de iones que conducen a la generación del “potencial de acción” cuya consecuencia es la transmisión del impulso eléctrico y la contracción mecánica de la célula. Sutiles cambios de base genética en la estructura molecular de estas proteínas (canalopatías) son las causantes de la disfunción de estos canales y de la consiguiente predisposición a la inestabilidad eléctrica en forma de diversas taquiarritmias.
El corazón es un órgano de función eléctrica y mecánica. La función mecánica de generar una presión arterial que haga circular la sangre por todo el organismo es la consecuencia de la contracción del miocardio. Esta contracción se deriva de una excitación eléctrica que se origina normalmente en un punto situado en la aurícula derecha (nodo sinusal) y que se transmite secuencialmente al resto de las estructuras cardíacas. Cuando las fibras cardíacas son excitadas por la llegada de ese impulso eléctrico el balance eléctrico celular (voltaje) entre el interior y el exterior de la célula se invierte y como consecuencia las fibras se acortan (se contraen) generando la presión mecánica para hacer circular la sangre. Este ciclo continuo de equilibrio eléctrico de excitación-contracción-relajación puede alterarse por un conjunto de disfunciones cuya base molecular es consecuencia de alteraciones en la estructura de esas proteínas que constituyen los canales iónicos. Tales disfunciones de los canales iónicos se derivan de sutiles cambios en la secuencia de aminoácidos de las proteínas que constituyen los canales producidos por mutaciones genéticas. Estas modificaciones moleculares alteran el flujo intra/extracelular de un ión determinado a través de la membrana, con escasa trascendencia funcional en base latido-latido, pero de gran impacto clínico cuando esa disfunción se ve magnificada puntualmente por un determinado desencadenante.
Los genes y la herencia en este contexto
El gen es la unidad de la herencia, una cadena molecular dentro de la larguísima secuencia de ADN de un cromosoma que se encarga de producir una proteína que tendrá una función concreta específica. En el contexto que nos ocupa se trata de las proteínas que constituyen los canales iónicos y/o la unión entre las células cardíacas para la transmisión sin retraso del impulso eléctrico en el miocardio. Las mutaciones son cambios aleatorios que se producen inadvertidamente en la estructura de un gen y que se transmiten a la siguiente generación al incorporarse a la dotación genética de sus descendientes. Tales mutaciones pueden ser beneficiosas o adversas pues la Naturaleza no es finalista en sí misma. Así, hace millones de años, una mutación en el gen que determina el color del pelaje de los osos que poblaban el Ártico hizo que éste fuera blanco y al conferirles esta mutación ventajas de no ser detectados por sus presas provocó que hoy todos los osos polares sean obviamente blancos. Las mutaciones en los canales iónicos implicados en los síndromes hereditarios arritmogénicos son todas ellas adversas pues producen inestabilidad eléctrica y facilitan el desarrollo de muerte prematura por arritmias ventriculares malignas. Hasta que dichas anomalías genéticas puedan editarse y corregirse para la próxima generación nuestra misión es detectar su existencia, estratificar el riesgo arrítmico y prevenir la muerte súbita.
Las QT-patías: el síndrome del QT largo
El síndrome del QT largo (SQTL) es la piedra Rosetta sobre la que se comenzó a descifrar la biología molecular de estas enfermedades. El intervalo QT del electrocardiograma es el sumatorio de la despolarización y repolarización del conjunto de células cardiacas ventriculares y su consecuencia mecánica de contracción sistólica ventricular. Es un parámetro muy constante, aunque su medida debe corregirse según la frecuencia cardíaca. Se acepta que el límite superior de ese intervalo corregido sea de 460 ms en mujeres y de 450 ms en hombres. La incidencia real de lo que en conjunto se conoce como QT-patías puede ser incluso superior a 1/2500-1/3000 personas ya que muchos sujetos no están identificados y que un tercio de los pacientes pueden tener (paradójicamente) en reposo un intervalo dentro de límites normales. Desde que en 1957 se describiera el primer caso de este síndrome, los avances en el conocimiento de su genética han posibilitado incluso la asociación genotipo-fenotipo en muchos casos, es decir la relación entre el mecanismo genético subyacente y sus manifestaciones electrocardiográficas y clínicas, lo que repercute enormemente sobre la farmacoterapia y sus aspectos preventivos y pronósticos.
Hoy día, más de 600 mutaciones de 17 genes diferentes se han asociado con el síndrome (LQT1-LQT17) cuya modificación afecta la funcionalidad de diversos canales iónicos. De relevancia clínica por su prevalencia son las tres primeras ya que representan más del 90% de los casos de SQTL (Figura 2).
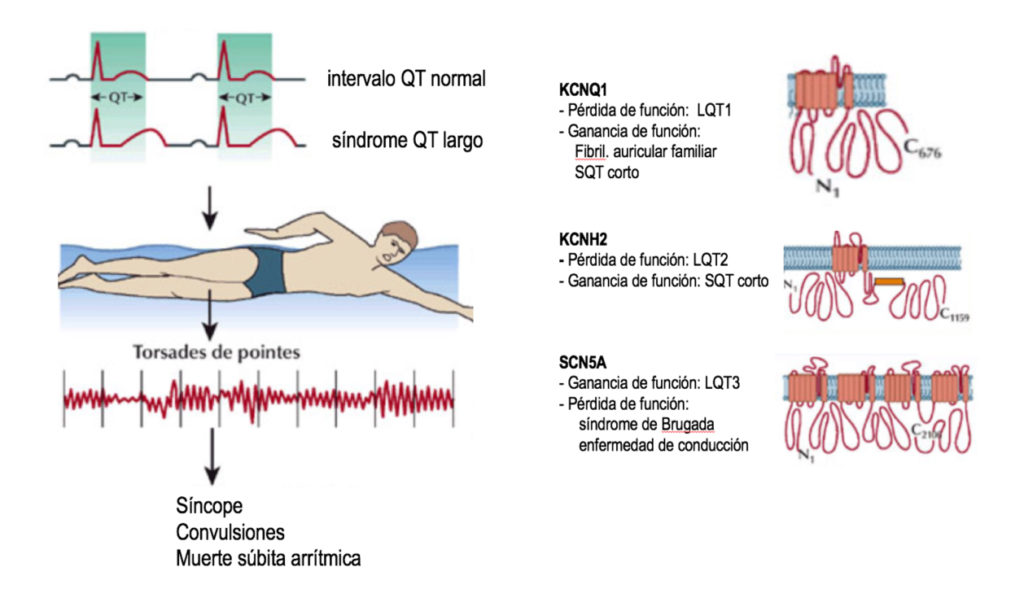
FIGURA 2. El síndrome de QT largo se caracteriza por una prolongación del intervalo QT del electrocar- diograma, que es el tiempo de “recarga” del conjunto de las células cardíacas tras una despolarización, pudiendo apreciarse en el ECG en reposo de estas personas sin consecuencia funcional directa. Sin embargo, un desencadenante (como la natación especialmente en agua fría en el LQT1) puede puntualmente ocasionar una repentina desestabilización del equilibrio eléctrico ventricular en forma de una arritmia paradigmática en este contexto denominada “Torsades de pointes” cuyas consecuencias pueden ser mortales si no se autolimita. Desde 1995 se han caracterizado las bases moleculares de muchas de estas canalopatías, habiéndose descrito cientos de mutaciones en un conjunto de genes específicos.
En el SQTL el “fallo técnico” que lo origina suele
relacionarse con la pérdida de función de un canal de potasio o ganancia de función del canal de sodio.
Así, el LQT1 se relaciona con una mutación en el gen KCNQ1 que representa un 40-55% de los individuos genéticamente positivos para el SQTL. Los eventos arrítmicos y sus consecuencias como síncope o muerte súbita en estos sujetos suelen manifestarse durante ejercicio/emociones y en particular durante la natación. El LQT2 se asocia con mutación en KCNH2 y representa entre 30-45% de los casos.
En este grupo son los estímulos auditorios repentinos (alerta, sonido despertador, trueno…) los causantes de las arritmias. Hace años, estudiamos a una mujer con este tipo LQT2 que describía perfectamente cuadros sincopales repentinos en la infancia mientras jugaba al escondite y era sorprendida (Figura 3).
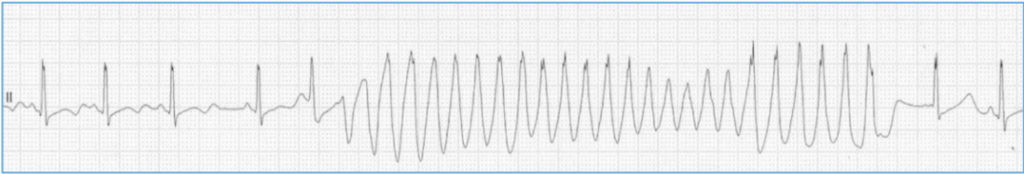
Figura 3 A. Ritmo Sinusal

Figura 3 B. Fibrilación Ventricular
FIGURA 3. En muchas canalopatías de base hereditaria como el síndrome QT largo las arritmias malignas se presentan en dos versiones como las aquí ilustradas. Arriba (A) un posible desencadenante (ejercicio, emoción, etc) inicia una taquiarritmia ventricular muy rápida y polimorfa (Torsade de Pointes) que dependiendo de su duración ocasionará presíncope o síncope brusco pero que finalmente se autolimita segundos después. Siendo repetitivos y de pronta recuperación, muchas veces no se presta atención a estos síncopes.
El principal riesgo es que eventualmente uno de tales episodios (B) puede no autolimitarse y continuar como fibrilación ventricular y causar la muerte -excepto si es portador de desfibrilador. Dependiendo de la patología causante se estima que entre 25-50% casos de muerte súbita por estos trastornos hereditarios se preceden (meses, años) de síncopes por taquicardias ventriculares polimorfas como el de la figura 1A pero que no han sido correctamente filiados
Milagrosamente había sobrevivido a muchas arritmias ventriculares malignas que solían autolimitarse en segundos. Esto no ocurrió, sin embargo, en el ejemplo que se comenta al inicio del artículo pues la arritmia supuestamente persistió causando la muerte. El LQT3 es bastante menos frecuente y representa el 5-10% de estos síndromes, presentando las arritmias generalmente en reposo. Cuando el intervalo QT es anormalmente corto (QTc < 330-350 ms) también la incidencia de diversas arritmias, incluyendo las malignas y muerte súbita es mayor, aunque la incidencia y detección de estos síndromes QT corto es muy baja. Un claro ejemplo de la forma diversa en que cualquier disrupción del equilibrio natural de los canales iónicos incrementa el riesgo de arritmias malignas es el hecho de que una pérdida de función del canal de potasio subyace en los tipos LQT1 y LQT2 mientras que a la inversa, la ganancia de función de dicha corriente origina el síndrome de QT corto, cuya capacidad arritmogénica es incluso mayor.
Desde el punto de vista clínico, es importante señalar que a veces los cuadros sincopales instantáneos que caracterizan estos síndromes han sido erróneamente diagnosticados de epilepsia, cuando en realidad tienen obviamente un origen arrítmico: una taquiarritmia ventricular polimorfa que se autolimita tras varios segundos (Torsades de pointes). El diagnóstico diferencial en ocasiones es difícil, máxime si tenemos en cuenta que ciertos tipos de epilepsia pueden asociarse o coexistir con trastornos eléctricos cardíacos primarios. En el momento actual la terapéutica de este conjunto de síndromes de QT largo se basa fundamentalmente en el uso de ciertos fármacos para mejorar la estabilidad eléctrica (betabloqueantes, especialmente propranolol y nadolol) o la prevención directa de muerte súbita mediante desfibrilador implantable tras la estratificación de riesgo de estos individuos. En este sentido, el denominado desfibrilador subcutáneo cuyo implante no requiere de un electrodo endovascular proporciona importantes ventajas respecto al dispositivo convencional en este grupo de población de edad más joven. Los estudios genéticos aportan ya valiosa información para dirigir la terapia y contribuirán a optimizar el manejo cada vez de forma más decisiva.
Ciertos fármacos tienen capacidad de prolongar el intervalo QT y provocar inestabilidad eléctrica en forma similar a las formas congénitas previamente descritas. Muchos de estos fármacos (antihistamínicos, etc.) han sido retirados del mercado por dicho motivo. Ciertos antiarrítmicos, como la amiodarona, prolongan ligeramente dicho intervalo, pero sin consecuencias proarrítmicas ya que esta sustancia de uso habitual actúa sobre diversos canales iónicos con efecto final de estabilización eléctrica. Sin embargo, ciertos individuos con un intervalo ECG normal o ligeramente prolongado son portadores de anomalías en la función de ciertos canales iónicos que no se han detectado y que pueden ponerse de manifiesto tras administrar sustancias como la amiodarona. Estas personas –frecuentemente mujeres- evidencian una baja reserva de repolarización y el fármaco puede llegar a producir arritmias ventriculares de compromiso vital. Esta paradoja denominada “proarritmia” de que un fármaco usado para tratar una arritmia “benigna” desencadene una arritmia potencialmente grave debe tenerse siempre presente al prescribir estos fármacos. En el futuro será fundamental detectar individuos con dicha susceptibilidad mediante test genético u otras vías. Mientras tanto, es primordial mantener un índice de sospecha de tal efecto y valorar el intervalo QT y otros parámetros del ECG tras administrar estos fármacos.
Síndrome de Brugada
En 1992 los hermanos Brugada describieron un patrón electrocardiográfico que se asociaba a muerte súbita arrítmica, caracterizado por una elevación convexa en precordiales derechas del segmento ST (Figura 4).
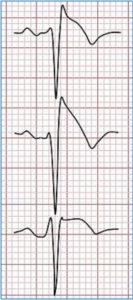
FIGURA 4. El patrón electrocardiográfico característico en precordiales derechas del síndrome de Brugada en sus manifestaciones más claras (Tipo 1) debe ser reconocido de inmediato por cualquier profesional de la salud. Si el paciente refiere en anamnesis algún cuadro sincopal brusco debe ser evaluado por un especialista con carácter urgente pues un síncope arritmogénico es predictor de muerte súbita
Hasta la fecha se han descrito mutaciones en 19 genes relacionadas con el referido fenotipo (ECG) de esos pacientes, con un modo de transmisión autosómico dominante. Aunque el gen clásicamente asociado con el síndrome es el SCN5A -que modula el canal de Na- lo cierto es que más del 65% de los probandos
(caso índice o individuo afectado) con este síndrome permanecen genéticamente indeterminados, tras estudio genético, lo que confirma su genética heterogénea. La prevalencia mundial estimada es de un 0,10%, y es posible que sea superior en áreas endémicas del sudeste asiático.
Las arritmias malignas ventriculares que caracterizan el síndrome se presentan habitualmente durante el reposo sobre todo en el sueño nocturno. Aunque la herencia es autosómica dominante, la proporción de síntomas es mucho mayor en el varón (6:1) y éstos se inician clásicamente en los 15-40 años. Aunque la mayoría de los pacientes con diagnóstico clínico son varones, todavía no se conoce cómo el sexo modula la manifestación de la enfermedad. Estas circunstancias, junto a la elevada incidencia en el sudeste asiático explica que en algunas zonas de Laos existiese la costumbre de que en familias con antecedentes de muerte súbita los varones durmiesen disfrazados de mujer para que la muerte no se fijara en ellos durante el sueño –el pensamiento mágico no es patrimonio de las sociedades tribales y lo vemos a diario en nuestro entorno.
No sólo existe el riesgo de arritmias ventriculares en el síndrome. Las arritmias supraventriculares como la fibrilación auricular se detectan en un 15-20% de los pacientes. También el bloqueo auriculoventricular y los retrasos de la conducción intraventricular forman parte del fenotipo del síndrome de Brugada. Desde hace años se reconoce que esta canalopatía se acentúa por el aumento de temperatura corporal (fiebre), que acentúa las manifestaciones del ECG y facilita el desarrollo de arritmias graves. En consecuencia, no es inusual, por tanto, que en muchos casos se detecte el patrón electrocardiográfico que define este síndrome al realizar un ECG en Urgencias en un paciente que acude con fiebre de cualquier origen.
Un aspecto crucial en este síndrome es la estratificación de riesgos y sus implicaciones. Las técnicas de estratificación de riesgo tienen una capacidad limitada de predicción, lo que dificulta el manejo de estos individuos, máxime considerando que más del 60% de los casos diagnosticados del síndrome de Brugada están asintomáticos y se detectan en una revisión rutinaria con ECG. En pocas enfermedades la diferencia entre “sobretratar” (colocar un desfibrilador preventivo a un individuo de bajo riesgo teórico) o “infratratar” (no colocarlo en otro de alto riesgo y por tanto que puede morir súbitamente al no estar protegido) es tan crucial.
Lamentablemente, un porcentaje elevado de los pacientes con el diagnóstico se encuentra en la categoría de riesgo intermedio y -como se ha repetido- nuestras herramientas de estratificación de riesgo son poco específicas. El estudio electrofisiológico y la estimulación eléctrica programada se utilizan habitualmente para este cometido, pero su capacidad predictiva de acontecimientos es poco específica en la mayoría de casos con resultados inconcluyentes. Para el clínico general es importante saber que un 25% de pacientes con este síndrome que experimentan una muerte súbita previamente habían referido algún cuadro sincopal brusco. Este dato obliga a una correcta anamnesis y a buscar el síncope de forma dirigida en la anamnesis, pues se confirma que es un marcador valioso en la predicción de muerte súbita.
En cuanto a la terapéutica, hasta la fecha ningún tratamiento médico ha resultado eficaz para prevenir las arritmias y la muerte súbita en el síndrome. El implante de desfibrilador es el único tratamiento eficaz, aunque en pacientes con descargas frecuentes por arritmias ventriculares recurrentes, aparenta ser muy útil la ablación epicárdica de potenciales específicos en tracto de salida de ventrículo derecho. Otra alternativa eficaz puede ser farmacológica: la quinidina, cuyo uso cabe considerarse para el tratamiento adyuvante en casos de arritmias recurrentes y descargas frecuentes del desfibrilador.
Resumen y perspectivas de esta Parte I
Pocas áreas de la Medicina tienen un potencial de desarrollo tan espectacular a medio y largo plazo como la Genética y la biología molecular. En unas décadas su influencia será decisiva en todos los campos de la Medicina tanto en su vertiente preventiva como en la terapéutica. El nivel de conocimiento actual de la biología molecular se atestigua, por ejemplo, en la asombrosa disponibilidad de una vacuna efectiva para el SARS-CoV-2 desarrollada en solo varios meses.
El campo de las arritmias cardíacas de base hereditaria viene experimentando un enorme desarrollo en los últimos años. La constante aportación de nueva información y datos en la genética de estas patologías junto a los avances en el diagnóstico y estratificación de riesgo acercan la posibilidad de un enfoque más personalizado e incluso un tratamiento “gen-específico” basado en el diagnóstico molecular. La identificación de mutaciones en los genes que causan estas enfermedades ha permitido un conocimiento más preciso en la fisiopatología.
Los estudios genéticos vienen cobrando una importancia creciente en estas patologías. Así, estos estudios han permitido establecer patrones electrocardiográficos gen-específicos, desencadenantes arritmogénicos gen-específicos, respuesta terapéutica a betabloqueantes gen-éspecíficos y estratificación de riesgo gen-especifica. Recurrir, pues, a la biología molecular para dicha finalidad resulta especialmente atractivo cuando las pruebas invasivas o no invasivas para la cuantificar el riesgo carecen de valor predictivo. En este sentido, conocer de antemano si una persona presenta una susceptibilidad genética p. ej. a prolongar su intervalo QT basalmente normal en presencia de un determinado fármaco o desencadenante particular tendría indudablemente un enorme interés preventivo, especialmente en ciertas familias.
En la actualidad, la información que brinda el estudio genético no es uniforme en todas las canalopatías, pero el ritmo de los avances en investigación traslacional augura un futuro esperanzador.
![]()