

Un policía municipal da el alto a un joven motorista para un control rutinario dentro de población. Cuando el agente pide la documentación el chico sufre un colapso instantáneo del que se recupera segundos después. Se asiste en Centro de Salud donde todas las pruebas son normales. Acude a consulta donde se constata que en la familia hay antecedentes de muerte súbita. Tras varias pruebas y bajo sospecha de taquicardia ventricular catecolaminérgica se implanta un desfibrilador.
Aproximadamente un año después, mientras montaba en una atracción de feria, sufre una parada cardíaca repentina motivada por una fibrilación ventricular, recuperándose tras descarga del desfibrilador. A lo largo de los últimos 15 años ocasionalmente ha presentado alguna descarga por fibrilación ventricular siempre en condiciones de estrés mental o físico.
Taquicardia ventricular catecolaminérgica
El caso ilustrado es un ejemplo paradigmático de taquicardia ventricular polimorfa catecolaminérgica (TVPC) a la par que representativo de las enfermedades hereditarias de arritmias cuya segunda parte se aborda en este artículo. Dos tías carnales del caso referido habían fallecido súbitamente y un primo hermano (hijo de una de aquellas) sufrió una parada cardíaca en su infancia de la que pudo ser reanimado y es también actualmente portador de desfibrilador.
La taquicardia ventricular polimórfa catecolaminérgica tiene una prevalencia estimada en 1/10,000. Se caracteriza por arritmias ventriculares en contexto adrenérgico (ejercicio, estrés) que puede resultar en síncope (si la arritmia se autolimita en breves segundos) o muerte súbita (si la arritmia persiste y desarrolla fibrilación ventricular). Tal comportamiento es frecuente en este tipo de síndromes arrítmicos hereditarios, tal como se comentó en la primera parte de este artículo. La TVPC fue descrita inicialmente en 1978, siendo relacionada años más tarde su fisiopatología con la homeostasis del calcio. La TVPC se engloba dentro de las denominadas canalopatías, y es una de las formas más severas entre los trastornos arritmogénicos hereditarios.
Desde punto de vista genético, Silvia Priori identificó en 2001 el receptor cardiaco de la rianodina (RyR2) como el primer gen asociado a la forma autosómica dominante de la TVPC. Dicho receptor regula la liberación del calcio al citoplasma desde el retículo sarcoplásmico durante la fase de meseta del potencial de acción. Se han encontrado mutaciones en el RyR2 en el 60 % de los pacientes afectados clínicamente, habiéndose descrito más de 70 mutaciones. Menos frecuentemente, la mutación proviene del gen que codifica la calsequestrina (CASQ2), una proteína reguladora del calcio en el retículo sarcoplásmico, y que participa modulando la respuesta del receptor rianodínico al calcio intracelular. Ambos tipos de mutaciones resultan en una sobrecarga plasmática de calcio durante la diástole, que aumenta por la acción de las catecolaminas circulantes (ejercicio/emoción), y que por un mecanismo de post-despolarizaciones tardías y actividad desencadenada originan las arritmias ventriculares que caracterizan el síndrome.
En el diagnóstico de la TVPC resulta fundamental la clínica y la historia familiar. Hasta un 30 % de los pacientes presentan antecedentes familiares de síncope y de muerte súbita. El electrocardiograma (ECG) basal de reposo y el ecocardiograma suelen ser totalmente normales. Generalmente, la prueba de esfuerzo constituye la prueba diagnóstica esencial, ya que en ella se documentan con frecuencia arritmias ventriculares características, tanto no sostenidas como sostenidas.
Clásicamente, las arritmias ventriculares se van acentuando conforme aumenta la carga adrenérgica, iniciándose habitualmente a partir de frecuencias de 110-130 lpm cuando pueden observarse extrasístoles ventriculares aislados, y, posteriormente dobletes, bigeminismo, y taquicardias ventriculares habitualmente con morfología característica y alternancia en su eje en ECG (Figura 1).
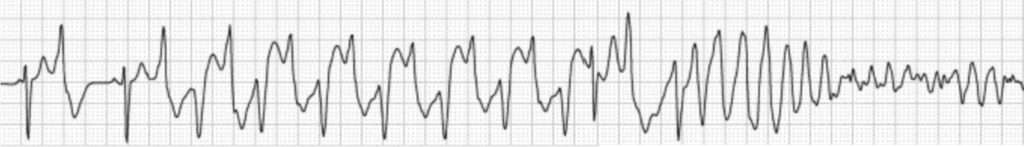
FIGURA 1. Taquicardia ventricular polimorfa catecolaminérgica (TVPC) durante una prueba de esfuerzo. La extrasistolia ventricular se va haciendo más frecuente y de comportamiento y morfología típicos en esta entidad hasta que finalmente desarrolla fibrilación ventricular. Cualquier situación adrenérgica (p. ej. de base emocional) puede tener la misma patogénesis y consecuencias en estos pacientes.
En ocasiones la ergometría puede ser inconcluyente, sin arritmias específicas al esfuerzo, lo que no garantiza ausencia de eventos en el seguimiento.
Clínicamente, las arritmias pueden aparecer ya en la infancia, y sin tratamiento se ha documentado una mortalidad de un 30 % en las tres primeras décadas de la vida en pacientes sintomáticos. Raramente, la muerte súbita puede ser la primera manifestación de la enfermedad. Los fármacos betabloqueantes constituyen el tratamiento de elección. Sin embargo, y aunque su uso se asocia con tasas más bajas de eventos, estos no aseguran la desaparición de las arritmias ni de sus consecuencias potencialmente letales motivo por el que generalmente su terapéutica combina el implante preventivo de un desfibrilador con el uso de fármacos antiadrenérgicos. Aquí, nuevamente, resulta crucial la estratificación de riesgo para identificar los pacientes en quienes el tratamiento con betabloqueantes representa sólo una protección parcial. En tal sentido, dentro de los fármacos antiadrenérgicos el nadolol y el propranolol son los fármacos más eficaces.
Fibrilación ventricular idiopática
En un 5-15% de supervivientes de una parada cardiaca extrahospitalaria no se detecta -incluso tras estudio exhaustivo- la causa directa o indirecta de fibrilación ventricular (FV). Suele tratarse de individuos entre 20-50 años sin patologías previas y ECG anodino (Figura 2).
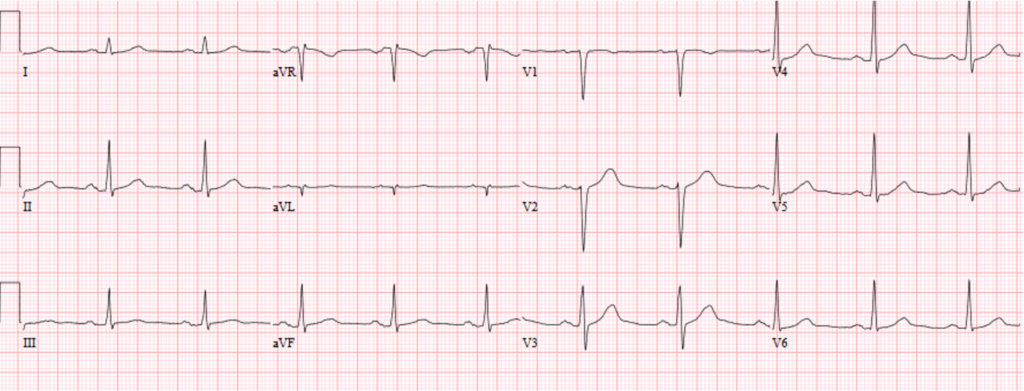
FIGURA 2. ECG de un varón de 38 años con fibrilación ventricular idiopática, reanimado con éxito de una parada cardíaca y actualmente portador de desfibrilador automático. El registro es rigurosamente normal, como lo fueron todas las pruebas complementarias practicadas.
Se agrupan bajo esta denominación aquellos casos de FV cuya causa sigue sin intuirse tras excluir un aparente origen cardíaco (estructural o eléctrico reconocido), respiratorio, metabólico, o tóxico. La mayoría de estos casos parecen relacionarse con un síndrome arrítmico de origen genético que predispone al desarrollo de extrasístoles ventriculares de acoplamiento corto. Los genes implicados hasta la fecha en esta entidad son el DPP6, el CALM1 (que codifica calmodulina) y el RYR2. El mecanismo arritmogénico exacto para explicar la forma en que la sobrexpresión de DPP6 causa la FV idiopática es incierto. Muchos de estos pacientes tras sobrevivir a la parada reciben un desfibrilador y –a veces años después- cuando presentan una recurrencia de la FV correctamente tratada por el dispositivo se observa en los electrogramas almacenados del episodio que el inicio de la FV se debe a una extrasístole ventricular de acoplamiento corto (Figura 3).
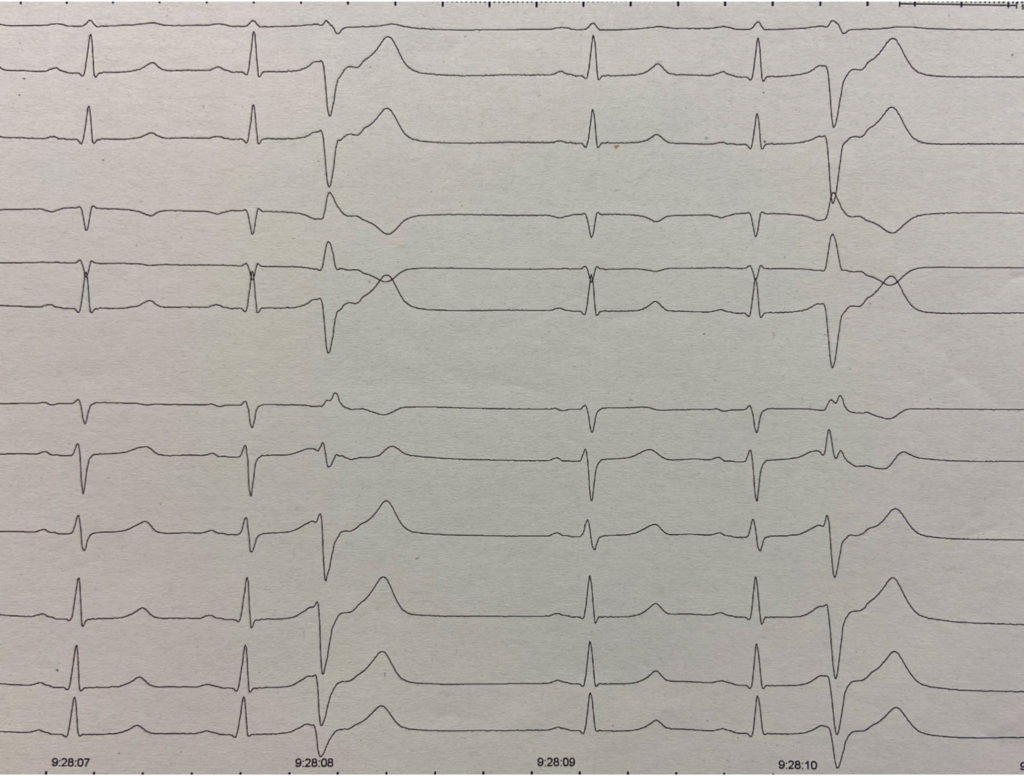
FIGURA 3. ECG de una mujer con fibrilación ventricular (FV) idiopática relacionada con el ocasional desarrollo de extrasistolia ventricular de muy corto acoplamiento: el extrasístole se inscribe en el inicio del descenso de onda T. Tras sobrevivir a la parada cardíaca se implantó un desfibrilador. La extrasistolia es muy errática en su comportamiento, desapareciendo completamente durante meses, lo que impide su mapeo preciso y ablación. En los últimos años ha presentado recurrencia de FV en dos ocasiones. En ambas la arritmia sostenida se precede de algún extrasístole aislado -como el señalado en este ECG obtenido horas después del episodio- hasta que finalmente uno de esos extrasístoles desencadena la fibrilación ventricular correctamente sensada y tratada por el dispositivo.
En algunos casos el origen de la extrasistolia se ha localizado en la banda moderadora del ventrículo derecho –un haz muscular que conecta el septo y la pared libre del ventrículo caracterizada por abundante distribución de fibras Purkinje. Pasado el evento es frecuente que la monitorización prolongada posterior no evidencie extrasistolia alguna. Aunque las extrasístoles ventriculares que actúan como desencadenantes suelen originarse en el tracto de salida del ventrículo derecho o en las fibras de Purkinje, resulta fundamental distinguir esta extrasistolia de la habitual de origen en el tracto de salida de ventrículo derecho cuyo comportamiento y consecuencias son completamente diferentes.
No hay datos fiables respecto a la utilidad de los fármacos en este síndrome de FV idiopática. Datos clínicos preliminares (coincidentes con nuestra propia experiencia) apoyan la utilidad de la quinidina: un fármaco clase IA con una actividad inhibitoria potente sobre la corriente Ito. Los pacientes que sobreviven a una parada cardíaca por FV idiopática deben recibir un desfibrilador implantable, reservando la terapia farmacológica con quinidina para casos de recurrencia frecuente de FV. En tales casos, el mapeo y ablación de las extrasístoles de corto acoplamiento que desencadenan la arritmia es una excelente opción si bien la eficacia de la técnica puede estar limitada por no poder reproducir la extrasistolia en el procedimiento, algo que ocurre con frecuencia.
Miocardiopatía hipertrófica
La miocardiopatía hipertrófica (MCH) es la patología cardiovascular hereditaria más frecuente, siendo estimada su prevalencia de 1:500 adultos. No es una canalopatía ni se trata de un trastorno eléctrico primario, pues existe una alteración estructural que, de hecho, da el nombre a la enfermedad. Sin embargo, su base genética e importancia como primera causa de muerte súbita en individuos jóvenes justifica su breve inclusión aquí. En 1990 se identificó la primera mutación genética ligada a la MCH familiar en un gen que codificaba la cadena pesada de la β-miosina (MYH7). Actualmente se reconoce la MCH como una enfermedad genética que afecta a las proteínas componentes del sarcómero cardiaco o de los discos Z, con un patrón de herencia autosómico dominante.
La MCH es familiar en la mitad de los casos y esporádica en la otra mitad. Tales casos esporádicos pueden darse por mutaciones nuevas (surgidas de novo) en el caso índice y que no estaban en sus progenitores. Algunos casos son falsos esporádicos: no han sido detectados en la generación previa por mostrar en ella una incompleta penetrancia.
Desde punto de vista arrítmico la principal amenaza de la MCH es la muerte súbita secundaria a arritmias ventriculares. Factores anatomopatológicos ligados a la MCH como la fibrosis miocárdica, la desorganización de los miocitos cardiacos (disarray) o la presencia de isquemia miocárdica por afectación microvascular predisponen a tales arritmias ventriculares. Además, la fibrilación auricular afecta a más del 20% de pacientes con MCH y se asocia a un mayor riesgo de ictus e insuficiencia cardiaca.
La MCH es una enfermedad compleja y clínicamente heterogénea donde la estratificación de riesgo es difícil en el paciente asintomático. Para establecer una correcta prevención primaria deben tenerse presentes un conjunto de factores de riesgo: la historia familiar de muerte súbita, sobre todo si la muerte ha ocurrido a edad temprana; el síncope cuya anamnesis sugiera posible origen arrítmico; la detección en Holter de taquicardias ventriculares no sostenidas, aunque es un hallazgo poco específico; la hipertrofia ventricular izquierda masiva (grosor septal ≥ 30 mm); la respuesta anómala de la presión arterial al ejercicio. Adicionalmente hay una serie de criterios cuya importancia va afianzándose en últimos años como la presencia de aneurismas apicales en ventrículo izquierdo o una extensa captación de gadolinio en la cardioresonancia entre otros.
La única estrategia capaz de prolongar la vida y alterar la historia natural de la MCH es el implante de un desfibrilador. En nuestra propia serie del Hospital Universitario Miguel Servet hemos observado como algunos pacientes con MCH y factores de riesgo en quienes se había implantado un desfibrilador de modo preventivo pueden tardar años en desarrollar síntomas, pero con frecuencia eventualmente acaba salvándoles la vida al materializarse tarde o temprano el riesgo de FV. La amiodarona es un antiarrítmico insuficiente por sí mismo en la prevención de muerte súbita en estos pacientes, pero resulta de utilidad como tratamiento complementario del desfibrilador para disminuir la carga arrítmica.
Miocardiopatía arritmogénica del ventrículo derecho
Habitualmente denominada como displasia de ventrículo derecho (DAVD). Como sucede en la MCH antes mencionada, en esta patología de nuevo existe unos cambios estructurales en el ventrículo –principalmente derecho- que definen la enfermedad, pero son sus consecuencias arrítmicas y su base genética las particularidades que justifican su inclusión en esta revisión. Es una entidad que se caracteriza por la sustitución progresiva de los miocitos normales por tejido graso y fibroso, predominantemente en el ventrículo derecho (VD), y que se asocia a taquicardias ventriculares (TV) y muerte súbita en ausencia de cardiopatía isquémica. El término de “displasia” fue propuesto tras observar la gran cantidad de grasa epicárdica e intramiocárdica presente en el VD sugerente de un defecto en el desarrollo del miocardio en dicho ventrículo.
La prevalencia de la DAVD varía según la zona geográfica estudiada, desde 1/2000 hasta 1/10000. En las zonas donde se ha llevado a cabo un mayor estudio de la enfermedad, como Francia o norte de Italia, se estima una prevalencia de 1/5000. Tiene una transmisión autosómica dominante, con grados diferentes de penetrancia y expresión incompleta, soliendo existir una historia familiar en el 30-70% de los casos. Los estudios de genética molecular han demostrado que la DAVD es una cardiopatía “desmosomal” resultado de defectos en las proteínas de adhesión celular como la placoglobina, desmoplaquina, y otras.
La DAVD es una entidad caracterizada por un deterioro estructural lentamente progresivo de miocardio afectando especialmente al ventrículo derecho. Estos cambios estructurales generan un substrato propicio para la aparición de diferentes arritmias a lo largo de la evolución de la enfermedad. La primera manifestación puede ser una muerte súbita, típicamente en jóvenes, pero también TV sostenidas y no sostenidas, con diferentes morfologías y con diferentes grados de tolerancia hemodinámica, así como extrasistolia ventricular frecuente.
Como el ventrículo izquierdo suele estar básicamente indemne y funcionalmente estos individuos suelen encontrarse asintomáticos la principal limitante para el diagnóstico de DAVD deriva en ocasiones de no haberse considerado en el diagnóstico diferencial de extrasistolia ventricular o de algunos tipos de taquicardia ventricular. Diversos estudios sugieren que en España entre un 5-15% de las muertes súbitas en sujetos de menos de 30 años pueden deberse a fibrilación ventricular relacionada con la existencia de DAVD –en muchos casos no identificada previamente y a menudo en deportistas. Mantener un adecuado índice de sospecha resulta pues fundamental para identificar un posible caso de DAVD. Las sospechas de esta patología deben plantearse en presencia de jóvenes con taquicardias QRS ancho con morfología de bloqueo de rama izquierda -ya que suelen originarse de áreas displásicas de ventrículo derecho-, síncopes presumiblemente arrítmicos durante/post ejercicio. En las pruebas complementarias: ecocardiograma con alteraciones morfológicas sutiles en ventrículo derecho, y en ECG basal discretas melladuras en QRS y/o la presencia de alteraciones de repolarización tipo ondas T invertidas en precordiales derechas.
En el entrenamiento de ejercicio de elevada intensidad y resistencia el ventrículo derecho es la estructura que soporta un especial sobreesfuerzo ya que la durante el ejercicio las resistencias pulmonares descienden mucho menos que las sistémicas, que son afrontadas por el ventrículo izquierdo. Probablemente éste sea un factor que justifique la aparente mayor incidencia de formas displásicas de ventrículo derecho en individuos con alto nivel de entrenamiento de resistencia (ciclistas de gran fondo, por ejemplo). Probablemente, estos sujetos albergan cierta predisposición genética con escasa penetrancia, pero el sobre entrenamiento desenmascara dicha susceptibilidad y promueve las alteraciones anatómicas y funcionales del ventrículo derecho.
La gran diversidad de eventos arrítmicos resultado de lo extenso y progresivo de la degeneración del miocardio resulta en que el tratamiento médico o la ablación con catéter sean paliativos proporcionando una eficacia limitada. En este sentido, es fundamental identificar a aquellos sujetos de alto riesgo donde el implante de un desfibrilador será imprescindible. El diagnóstico preciso y el manejo de tales pacientes debe ser establecido en las Unidades de Arritmias.
Enfermedad progresiva de conducción cardíaca
Se trata de un defecto de la propagación del impulso a través del sistema His-Purkinje (también se le conoce como enfermedad de Lev-Lenegre). Suele manifestarse inicialmente como un bloqueo de rama izquierda o derecha y avanza progresivamente hasta desarrollar un bloqueo completo. Habitualmente están implicadas mutaciones del SCN5A, siendo una de las causas más frecuentes de trastornos de conducción en ausencia de cardiopatía estructural.
Fibrilación auricular familiar
La fibrilación auricular (FA) es la arritmia más prevalente en la población general con una incidencia que aumenta con la edad hasta superar el 10% en la octava década. Habitualmente se asocia con diversas cardiopatías y factores de riesgo cardiovasculares: hipertensión, valvulopatías, cardiomiopatías, etc. El deporte de resistencia de alto nivel practicado desde la juventud también se asocia con la predisposición a desarrollar FA en los varones –curiosamente esta asociación no se ha observado en las atletas. En aproximadamente un tercio de casos de FA sin factores de riesgo para su desarrollo existen antecedentes familiares de fibrilación auricular. La genética de la fibrilación auricular familiar es compleja, habiéndose relacionado principalmente con defectos de función de varios canales de potasio.
Síndrome Wolff-Parkinson-White de presentación familiar
La mayoría de casos de preexcitación ventricular o síndrome de Wolff-Parkinson-White (WPW) en su presentación clásica carecen de una asociación familiar. Sin embargo, mientras que la incidencia del síndrome es del 0,15% en la población general se ha demostrado que la incidencia aumenta al 3,4% en familiares de primer grado de los pacientes con WPW. Hasta la fecha varias familias con este síndrome han sido estudiadas, habiéndose identificado una mutación en el gen PRKAG2 como causante. Independientemente de la base genética que la justifique, la asociación familiar es evidente como en el caso que se muestra en la Figura 4 que muestra dos hermanos gemelos, ambos exhibían patrón de preexcitación ventricular muy similar.
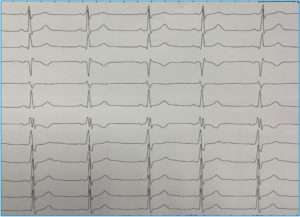
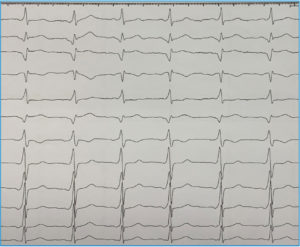
FIGURA 4. ECG de dos hermanos varones gemelos. Ambos presentaban síndrome de Wolff-Parkinson-White. Fueron sometidos a ablación. En (A) la vía se localizaba en punto posterolateral izquierdo del anillo mitral y en (B) era posteroseptal, tan solo unos 15 mm más septal que su hermano. Resulta llamativa la asombrosa similitud fenotípica de ambos hermanos incluso en heredar el defecto del desarrollo del aislamiento entre aurícula y ventrículo que representan las vías accesorias AV.
![]()